



















































Stwardnienie rozsiane – objawy i skuteczna walka z chorobą
15 maja 2019,
dodał: Redakcja
dodał: Redakcja
Artykuł zewnętrzny

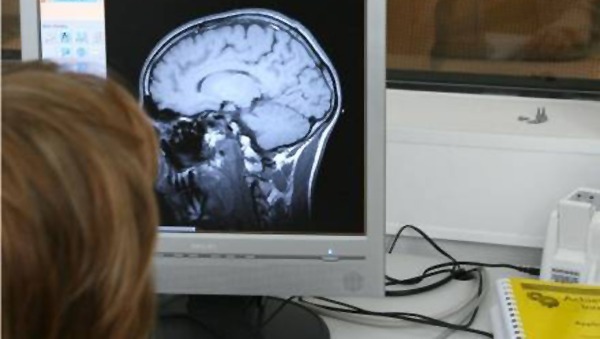
Stwardnienie rozsiane (SM) jest jedną z najczęstszych przewlekłych i nieuleczalnych chorób neurologicznych, centralnego układu nerwowego, która na ogół diagnozowana jest między 20-tym a 40-tym rokiem życia, ale coraz częściej także u osób poniżej 18-ego roku życia oraz u dzieci, będąca jednocześnie główną przyczyną inwalidztwa w grupie młodych ludzi. Szacunkowo na świecie choruje około 2,5 mln osób, z tego ok. 500 tys. w Europie. Największa zachorowalność występuje wśród ludzi rasy białej żyjących w klimacie umiarkowanym. Kobiety chorują częściej niż mężczyźni: na 10 osób z SM, 6 stanowią kobiety. W Polsce szacunkowo żyje ok. 50-60 tys. chorych na SM (1 osoba chora na ok. 600 obywateli). Mimo szybkiego postępu wiedzy i nadziei związanych z wprowadzeniem w ostatnich dwóch dekadach preparatów interferonu beta i octanu glatirameru, nadal nie ma istotnego przełomu w leczeniu tej choroby.
Pierwsze wzmianki, dotyczące objawów stwardnienia rozsianego pojawiły się w średniowieczu, a dokładnie w notatkach Jana Gerlacusa, który opisał chorobę św. Lidwiny z Schiedam (1380-1433). Pierwszy makroskopowy obraz plak demielinizacyjnych został przedstawiony w 1828 roku w Londynie przez lekarza patologa, Roberta Hoopera. Z odkryciem SM łączy się jednak głównie nazwisko ojca nowoczesnej neurologii, Jeana-Marie Charcota, który zauważone przez siebie schorzenie to nazwał sclerose en plaques disseminees, tzn. twarde rozproszone blizny. Badania nad SM prowadzono również w Polsce, gdzie po raz pierwszy w 1848 roku we Wrocławiu, F. Frerich postawił rozpoznanie sclerosis multiplex.
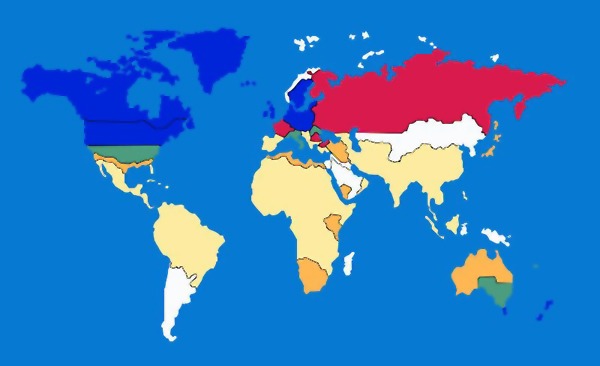
Badania geograficznego rozmieszczenia stwardnienia rozsianego wykazały wyraźną zależność zapadalności i umieralności na SM od szerokości geograficznej. Choroba występuje głównie między 44 a 64 stopniem szerokości geograficznej, a w pobliżu równika jest rzadkością. W związku z tym, można wyróżnić trzy obszary ryzyka zachorowań:
1) obszar wysokiego zagrożenia chorobą, o współczynniku rozpowszechnienia choroby powyżej 40/100 000/rok obejmuje: kraje bałtyckie, północno-zachodnią część Rosji, prawie całą Europę wraz z Polską, północne Włochy, południową Kanadę, północną część Stanów Zjednoczonych, południową Australię, Nową Zelandię i Tasmanię,
2) obszar umiarkowanego zagrożenia, chorobowość powyżej 20-39/100 000/rok. Należą do niego: południowo-zachodnia Norwegia, Laponia, południowa Ukraina, Kazachstan, zachodnia Syberia, przybrzeżne regiony Morza Śródziemnego (wyłączając Albanię, Korsykę, Tunis i Izrael), południowe Stany Zjednoczone Ameryki Północnej, Argentyna, Hawaje, północna Australia i Afryka Południowa,
3) obszar niskiego zagrożenia, rozpowszechnienie choroby poniżej 20/100000/rok. Zalicza się do niego: prawie wszystkie kraje Afryki, Azję, państwa karaibskie, Oceanię, Meksyk.
1) obszar wysokiego zagrożenia chorobą, o współczynniku rozpowszechnienia choroby powyżej 40/100 000/rok obejmuje: kraje bałtyckie, północno-zachodnią część Rosji, prawie całą Europę wraz z Polską, północne Włochy, południową Kanadę, północną część Stanów Zjednoczonych, południową Australię, Nową Zelandię i Tasmanię,
2) obszar umiarkowanego zagrożenia, chorobowość powyżej 20-39/100 000/rok. Należą do niego: południowo-zachodnia Norwegia, Laponia, południowa Ukraina, Kazachstan, zachodnia Syberia, przybrzeżne regiony Morza Śródziemnego (wyłączając Albanię, Korsykę, Tunis i Izrael), południowe Stany Zjednoczone Ameryki Północnej, Argentyna, Hawaje, północna Australia i Afryka Południowa,
3) obszar niskiego zagrożenia, rozpowszechnienie choroby poniżej 20/100000/rok. Zalicza się do niego: prawie wszystkie kraje Afryki, Azję, państwa karaibskie, Oceanię, Meksyk.


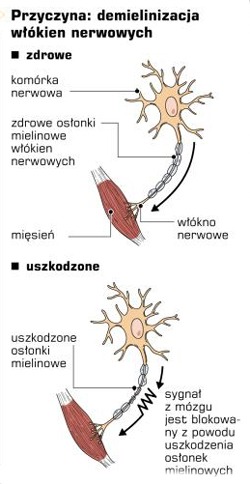
Stwardnienie rozsiane (łac. sclerosis multiplex, SM, ang. multiple sclerosis, MS) jest chorobą autoimmunologiczną. Oznacza to, że układ odpornościowy organizmu atakuje swoją własną tkankę w błędnym przekonaniu, iż jest to obce ciało. Z łaciny sclerosis oznacza stwardnienie, którego ogniska widoczne są w badaniu mózgu chorego, multiplex – rozsiane, gdyż ogniska te mogą pojawić się w wielu miejscach mózgu i rdzeniu kręgowym. W przypadku SM system immunologiczny atakuje mielinę, czyli substancję otaczającą komórki nerwowe w mózgu i rdzeniu kręgowym. Uszkodzenia osłonki mielinowej wokół nerwów powodują częściowe lub całkowite przerwanie przewodzenia impulsów nerwowych, co skutkuje upośledzeniem danej funkcji lub jej utratą. Mówiąc inaczej dochodzi do wieloogniskowego uszkodzenia (demielinizacji i rozpadu aksonów) tkanki nerwowej. Starsze, wcześniej powstałe ogniska mają dość zwartą konsystencję, są stwardniałe, zbliznowaciałe (w wyniku m.in. żywego odczynu komórek glejowych) w odróżnieniu od miękkich (z obecnością licznych komórek zapalnych) świeżo powstałych zmian. Istotą obrazu choroby jest występowanie rozsianych zmian demielinizacyjnych w ośrodkowym układzie nerwowym (OUN), czyli tzw. plak. Obszarami ośrodkowego układu nerwowego szczególnie dotkniętymi chorobą są sznury boczne i tylne (zwłaszcza w odcinku szyjnym i lędźwiowym) rdzenia kręgowego. Zmiany chorobowe dość często dotyczą także nerwu wzrokowego oraz obszarów wokół komór w mózgu. Zajęte też bywają szlaki nerwowe w śródmózgowiu, moście oraz móżdżku. Zmiany dotyczą też kory móżdżku oraz rdzenia kręgowego. Proces chorobowy przez dłuższy czas oszczędza komórkę nerwową i jej długie wypustki, aksony. Pierwotnie zajęte są dendryty, wypustki, za pomocą których komórki komunikują się między sobą. W późniejszym okresie zmiany chorobowe dotykają jednak także i aksony.
 Przyczyna SM nie została, jak dotąd, w pełni ustalona. Wiadomo, że choroba ma podłoże immunologiczne. Podkreśla się nadal rolę zakażeń wirusowych w inicjacji zmian w ośrodkowym układzie nerwowym. Szczególną rolę przypisuje się wirusom latentnym, tj. powodującym zakażenia bez widocznych następstw klinicznych (wirus latentny, po wtargnięciu do komórki, zagnieżdża się w jej jądrowym genomie i przez jakiś czas tkwi w nim uśpiony). Sprawców SM szuka się np. wśród wirusów opryszczki lub – ogólnie rzecz biorąc – wśród retrowirusów. Infekcja wirusowa prawdopodobnie powoduje pobudzenie układu immunologicznego i jego fałszywą odpowiedź skierowaną przeciw antygenom mieliny, która stanowi bardzo istotny składnik osłonki nerwów, umożliwiający ich prawidłowe funkcjonowanie. Odpowiedź ma charakter zapalny. Zapalenie toczy się wokół drobnych naczyń odżywiających istotę białą. Uczestniczą w nim typowe dla stanów zapalnych komórki: makrofagi, plazmocyty i limfocyty, zwłaszcza limfocyty T (wszystkie ich rodzaje i podtypy). To właśnie limfocytom T i wydzielanym przez nie cytokinom przypisuje się największą rolę w ostatecznym niszczeniu mieliny. Rozwojowi choroby mogą sprzyjać także czynniki środowiskowe. SM występuje częściej w klimacie umiarkowanym i chłodnym niż gorącym. Z drugiej strony wiadomo też, że stan przegrzania w wyniku nadmiernej ekspozycji na ciepło może nasilić objawy choroby. Jak pokazują statystki najwięcej zachorowań na SM odnotowuje się w mniej nasłonecznionych regionach. Badania pozwalają sądzić, iż niedobór witaminy D – powstającej w wyniku działania promieni słonecznych może przyczyniać się do rozwoju choroby. Badania kliniczne ujawniają, że przyjmowanie witaminy D jest związane z mniejszą zapadalnością na SR, jak również z mniejszą aktywnością choroby. Mechanizm, przez który witamina D wpływa na SR, jest związany z jej funkcjami immunologicznymi. Być może istnieje predyspozycja genetyczna do SM – obserwuje się bowiem rodzinne występowanie tego schorzenia (ok. 10%), ustalono związki choroby z układem zgodności tkankowej HLA. Ostatnio podkreśla się, że na częstsze występowanie SR wśród kobiet, poza czynnikiem genetycznym, mogą wpływać hormony – estrogeny, a zwłaszcza prolaktyna, wydzielana m.in. przez limfocyty, wywierająca istotny efekt immunostymulujący i modulujący odpowiedź immunologiczną. Powoduje ona również negatywną selekcję limfocytów B w czasie ich dojrzewania, wpływając na swego rodzaju promowanie autoreaktywnych limfocytów B, wzmaga odpowiedź na antygeny i miogeny oraz zwiększa produkcję immunoglobulin i autoprzeciwciał. Prawdopodobnie na rozwój choroby wpływa również dieta, wysokokaloryczna, bogata w tłuszcze i cukry sprzyja wystąpieniu SM, niskokaloryczna może zmniejszyć ryzyko zachorowania. Badania epidemiologiczne wykazały, że nienasycone kwasy tłuszczowe mają pozytywny wpływ na przebieg SR, natomiast przyjmowanie dużej ilości nasyconych kwasów tłuszczowych wiąże się z większą zapadalnością na tę chorobę. Przyjmuje się, że korzystna jest dieta ograniczająca ilość spożywanych tłuszczów, w której nienasycone kwasy tłuszczowe zastępują tłuszcze nasycone. Szczególne znaczenie przypisuje się kwasowi linolenowemu, którego korzystny wpływ wykazano w kilku małych badaniach klinicznych. Rola makro- i mikroelementów, antyoksydantów, różnych witamin czy oleju rybiego nie jest potwierdzona jednoznacznie. Badania naukowe udowodniły jedynie, że niedożywienie może zaostrzać przebieg SR. Nie bez znaczenia jest też profilaktyka osteoporozy, bardzo częstego powikłania powtarzanego leczenia kortykosteroidami. Dość często jest też podawany związek między stresem a pogorszeniem funkcjonowania czy wystąpieniem rzutu choroby. Zgłaszany jest bardzo często przez pacjentów i potwierdzany badaniami klinicznymi, chociaż należy zawsze uwzględnić fakt, że przewlekle chorzy często w sposób nadmierny wiążą pogorszenie z wpływem czynników zewnętrznych, a szczególnie ze stresem psychicznym.
Przyczyna SM nie została, jak dotąd, w pełni ustalona. Wiadomo, że choroba ma podłoże immunologiczne. Podkreśla się nadal rolę zakażeń wirusowych w inicjacji zmian w ośrodkowym układzie nerwowym. Szczególną rolę przypisuje się wirusom latentnym, tj. powodującym zakażenia bez widocznych następstw klinicznych (wirus latentny, po wtargnięciu do komórki, zagnieżdża się w jej jądrowym genomie i przez jakiś czas tkwi w nim uśpiony). Sprawców SM szuka się np. wśród wirusów opryszczki lub – ogólnie rzecz biorąc – wśród retrowirusów. Infekcja wirusowa prawdopodobnie powoduje pobudzenie układu immunologicznego i jego fałszywą odpowiedź skierowaną przeciw antygenom mieliny, która stanowi bardzo istotny składnik osłonki nerwów, umożliwiający ich prawidłowe funkcjonowanie. Odpowiedź ma charakter zapalny. Zapalenie toczy się wokół drobnych naczyń odżywiających istotę białą. Uczestniczą w nim typowe dla stanów zapalnych komórki: makrofagi, plazmocyty i limfocyty, zwłaszcza limfocyty T (wszystkie ich rodzaje i podtypy). To właśnie limfocytom T i wydzielanym przez nie cytokinom przypisuje się największą rolę w ostatecznym niszczeniu mieliny. Rozwojowi choroby mogą sprzyjać także czynniki środowiskowe. SM występuje częściej w klimacie umiarkowanym i chłodnym niż gorącym. Z drugiej strony wiadomo też, że stan przegrzania w wyniku nadmiernej ekspozycji na ciepło może nasilić objawy choroby. Jak pokazują statystki najwięcej zachorowań na SM odnotowuje się w mniej nasłonecznionych regionach. Badania pozwalają sądzić, iż niedobór witaminy D – powstającej w wyniku działania promieni słonecznych może przyczyniać się do rozwoju choroby. Badania kliniczne ujawniają, że przyjmowanie witaminy D jest związane z mniejszą zapadalnością na SR, jak również z mniejszą aktywnością choroby. Mechanizm, przez który witamina D wpływa na SR, jest związany z jej funkcjami immunologicznymi. Być może istnieje predyspozycja genetyczna do SM – obserwuje się bowiem rodzinne występowanie tego schorzenia (ok. 10%), ustalono związki choroby z układem zgodności tkankowej HLA. Ostatnio podkreśla się, że na częstsze występowanie SR wśród kobiet, poza czynnikiem genetycznym, mogą wpływać hormony – estrogeny, a zwłaszcza prolaktyna, wydzielana m.in. przez limfocyty, wywierająca istotny efekt immunostymulujący i modulujący odpowiedź immunologiczną. Powoduje ona również negatywną selekcję limfocytów B w czasie ich dojrzewania, wpływając na swego rodzaju promowanie autoreaktywnych limfocytów B, wzmaga odpowiedź na antygeny i miogeny oraz zwiększa produkcję immunoglobulin i autoprzeciwciał. Prawdopodobnie na rozwój choroby wpływa również dieta, wysokokaloryczna, bogata w tłuszcze i cukry sprzyja wystąpieniu SM, niskokaloryczna może zmniejszyć ryzyko zachorowania. Badania epidemiologiczne wykazały, że nienasycone kwasy tłuszczowe mają pozytywny wpływ na przebieg SR, natomiast przyjmowanie dużej ilości nasyconych kwasów tłuszczowych wiąże się z większą zapadalnością na tę chorobę. Przyjmuje się, że korzystna jest dieta ograniczająca ilość spożywanych tłuszczów, w której nienasycone kwasy tłuszczowe zastępują tłuszcze nasycone. Szczególne znaczenie przypisuje się kwasowi linolenowemu, którego korzystny wpływ wykazano w kilku małych badaniach klinicznych. Rola makro- i mikroelementów, antyoksydantów, różnych witamin czy oleju rybiego nie jest potwierdzona jednoznacznie. Badania naukowe udowodniły jedynie, że niedożywienie może zaostrzać przebieg SR. Nie bez znaczenia jest też profilaktyka osteoporozy, bardzo częstego powikłania powtarzanego leczenia kortykosteroidami. Dość często jest też podawany związek między stresem a pogorszeniem funkcjonowania czy wystąpieniem rzutu choroby. Zgłaszany jest bardzo często przez pacjentów i potwierdzany badaniami klinicznymi, chociaż należy zawsze uwzględnić fakt, że przewlekle chorzy często w sposób nadmierny wiążą pogorszenie z wpływem czynników zewnętrznych, a szczególnie ze stresem psychicznym.
SM przebiega inaczej u każdej chorej osoby. U niektórych charakteryzuje się okresami pogorszenia i poprawy funkcjonowania, u innych ma formę postępującą. Zawsze jednak czyni życie utrudnionym i nieprzewidywalnym.
W roku 1996 przyjęto obowiązujący do dzisiaj podział kliniczny choroby wg Lublina (Neurology 1996) na następujące typy:
1) SM rzutowo-remisyjne (RR relapsing-remitting). Najczęściej występująca postać choroby, która dotyczy ok. 60% chorych. Objawy występują przez pewien czas (dni, tygodnie, miesiące), a następnie stan neurologiczny ulega częściowej lub całkowitej poprawie. Stan pogorszenia zdrowia nazywany jest rzutem. Dochodzi do niego, kiedy komórki układu odpornościowego atakują osłonę włókna nerwowego w mózgu lub rdzeniu kręgowym, tworząc tym samym stan zapalny. Kiedy stan zapalny cofa się, objawy ustępują częściowo (zdarza się, że pozostanie potem pewne uszkodzenie) lub całkowicie znikają. Określa się to mianem remisji. Remisje mogą trwać zarówno kilka miesięcy, jak i lat.
2) SM wtórnie postępujące (SP secondary-progressive). Jest to typ SM, kiedy u osób z SM rzutowo-remisyjnym występuje trwałe pogorszenie stanu zdrowia przez co najmniej 6 miesięcy, bez względu na to, czy w tym czasie pojawiają się rzuty. Statystycznie u ok. 65% osób z SM rzutowo-remisyjnym po średnio 15 latach rozwija się postać wtórnie postępującą.
3) Łagodna postać SM (PP primary-progressive). W tej odmianie po jednym lub dwóch atakach następuje całkowity powrót do zdrowia i SM nie rozwija się. Jeżeli u osoby chorej po 10-15 latach stan zdrowia nie pogorszył się i ma ona minimalną niepełnosprawność lub jej brak, również można mówić o SM przebiegającym w sposób łagodny. Postać łagodna występuje u ok. 20% osób chorych.
4) SM pierwotnie postępujące (PR progressive-relapsing). Jest to forma SM dotycząca 10-15% osób chorych. Osoby z pierwotnie postępującym SM są na ogół diagnozowane w wieku 40 lat lub później. Podczas gdy w innych postaciach SM zmiany demielinizacyjne pojawiają się zarówno w mózgu jak i w rdzeniu kręgowym, w pierwotnie postępującym SM większość uszkodzeń ma miejsce w rdzeniu kręgowym. Osoby z pierwotnie postępującym SM nigdy nie mają wyraźnych rzutów lub remisji. Choroba rozpoczyna się od pojawienia się problemów, np. z poruszeniem, które się stopniowo pogłębiają.
 Coraz lepsze możliwości diagnostyczne i terapeutyczne umożliwiły wyodrębnienie nowego pojęcia klinicznego w diagnostyce stwardnienia rozsianego – tzw. CIS, czyli zespołu izolowanego klinicznie (Clinical Isolated Syndrom). Pojęcie to powstało po wprowadzeniu do diagnostyki badania metodą rezonansu magnetycznego. Ta nowa postać SR obejmuje przypadki, gdy obserwuje się występowanie jednego izolowanego objawu uszkodzenia układu nerwowego (np. zapalenie pozagałkowe nerwu wzrokowego z objawami upośledzenia widzenia). Występowanie takiego stanu klinicznego uruchamia proces diagnostyczny, w wyniku którego w badaniu metodą rezonansu magnetycznego stwierdza się obecność typowych zmian w obrazie (typowa lokalizacja, liczba, wielkość). Na podstawie dotychczasowych obserwacji coraz powszechniej uważa się, że powinno to już implikować rozważenie włączenia leczenia immunomodulującego.
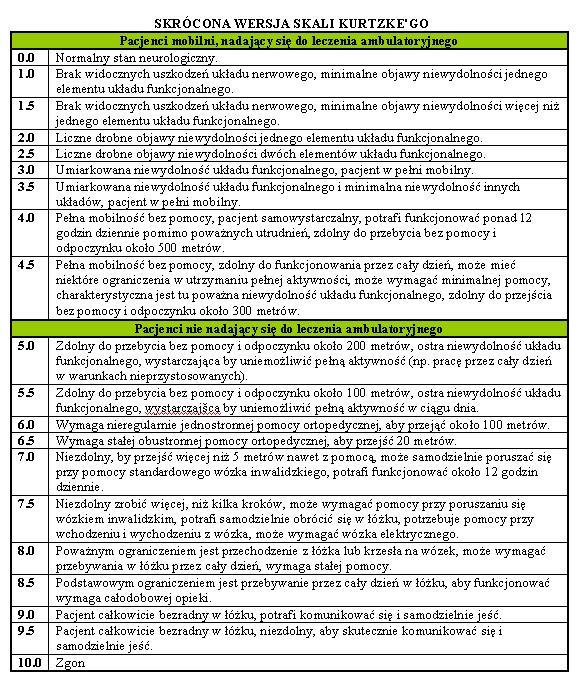
Coraz lepsze możliwości diagnostyczne i terapeutyczne umożliwiły wyodrębnienie nowego pojęcia klinicznego w diagnostyce stwardnienia rozsianego – tzw. CIS, czyli zespołu izolowanego klinicznie (Clinical Isolated Syndrom). Pojęcie to powstało po wprowadzeniu do diagnostyki badania metodą rezonansu magnetycznego. Ta nowa postać SR obejmuje przypadki, gdy obserwuje się występowanie jednego izolowanego objawu uszkodzenia układu nerwowego (np. zapalenie pozagałkowe nerwu wzrokowego z objawami upośledzenia widzenia). Występowanie takiego stanu klinicznego uruchamia proces diagnostyczny, w wyniku którego w badaniu metodą rezonansu magnetycznego stwierdza się obecność typowych zmian w obrazie (typowa lokalizacja, liczba, wielkość). Na podstawie dotychczasowych obserwacji coraz powszechniej uważa się, że powinno to już implikować rozważenie włączenia leczenia immunomodulującego.Najbardziej rozpowszechnioną i najszerzej akceptowaną skalą kliniczną jest tzw. EDSS (Expanded Disability Status Scale) opracowana przez Kurtzkego (Neurology 1983). W skali tej dokonuje sie oceny ośmiu tzw. systemów funkcjonalnych (Functional Systems FS) odpowiadających głównym funkcjom ośrodkowego układu nerwowego. Są to: motoryka, funkcje móżdżkowe, funkcje pnia mózgu, czucie powierzchowne i głębokie, funkcje pęcherza i jelita grubego, wzrok, funkcje mentalne, inne zaburzenia. Zestawienie i podsumowanie ilości punktów przyznanych przez badającego w każdym z ośmiu układów pozwala na zakwalifikowanie pacjenta wg skali od 0 do 4.0 punktów. Powyżej 4.0 punktów brane są pod uwagę głownie jego zdolności lokomocyjne, a zwłaszcza odcinek, który pacjent jest w stanie pokonać sam lub z pomocą. Fakt nadmiernej akcentacji tych zdolności ruchowych należy wg krytyków skali EDSS do jej głównych słabości. Drugą słabością ograniczającą statystyczną wagę tej metody opisywania stanu klinicznego jest jej nielinearność (dwa szczyty, jeden poniżej 4 punktów i jeden większy powyżej 6 punktów), wynikające z powyżej wymienionych założeń. Mimo tych zastrzeżeń EDSS jest ciągle jeszcze jedynym jednolitym mechanizmem klasyfikacji stanu chorego i dlatego właśnie każde badanie neurologiczne chorego na SM powinno umożliwić zapis jego wyniku w formie punktów EDSS. Skalą konkurencyjną dla skal Kurtzkego jest The Scripps Neurologic Rating Scale (SNRS), opracowana przez zespół Jamesa Koziola z Instytutu Badawczego Scrippsa w La Jolla w Kalifornii.
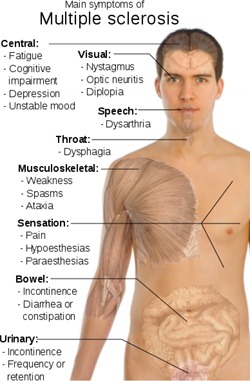
Choroba z reguły pojawia się dość nagle, zaskakuje chorego, który dotychczas był sprawną osobą, i przejawia się upośledzeniem lub utratą określonej czynności układu nerwowego. U każdego chorego na SM mogą pojawić się jednak inne objawy, które są uzależnione od miejsca wystąpienia ognisk choroby w układzie nerwowym.
Do najczęstszych objawów zaliczyć można:
1) uczucie zmęczenia, które jest nieprzewidywalne lub niewspółmierne do wykonywanej czynności,
2) zaburzenia wzroku – niewyraźne widzenie, widzenie podwójne, zapalenie nerwu wzrokowego, oczopląs, ból przy poruszaniu gałką oczną,
3) problemy z utrzymaniem równowagi i koordynacją ruchów – utrata równowagi, drżenie kończyn, niestabilny chód, zawroty głowy, utrata częściowej kontroli nad kończynami, brak koordynacji ruchów, uczucie osłabienia mięśni, niedowłady,
4) zaburzenia napięcia mięśniowego – spastyczność, wzmożone napięcie mięśniowe i sztywność mięsni, skurcze,
5) zmiany w odbiorze bodźców – uczucie mrowienia, drętwienia, palącego gorąca, ból o charakterze przewlekłym, np. nerwoból trójdzielny, bóle pozostałych mięśni,
6) zaburzenia mowy – mowa skandowana, spowolniona, bełkotliwa, zmiana rytmu mowy, trudności z połykaniem,
7) zaburzenia funkcji pęcherza moczowego i jelit – potrzeba częstszego wypróżniania się, niepełne wypróżnianie się, utrata kontroli nad momentem wypróżnienia, zaparcia,
8) zaburzenia w sferze – impotencja, zmniejszenie popędu płciowego, zmniejszenie wrażliwości na dotyk i inne bodźce,
9) nadwrażliwość na ciepło,
10) zaburzenia percepcji i zaburzenia emocjonalne – u trata pamięci krótkotrwałej, trudności z koncentracją, oceną sytuacji lub logicznym myśleniem, obniżenie nastroju, depresja.
Liczne badania kohortowe przeprowadzane w wielu ośrodkach i na dużych grupach chorych pozwoliły ustalić kliniczne, biochemiczne i rezonansowe wskaźniki progresji choroby. Mimo że żaden z tych czynników nie pozwala w sposób pewny i jednoznaczny prognozować przebiegu choroby, to wyodrębniono czynniki kliniczne związane z dobrym i złym rokowaniem. Dobre rokowanie jest związane z płcią żeńską, wczesnym początkiem choroby, zapaleniem nerwu wzrokowego lub objawami czuciowymi, jako pierwszymi objawami, postacią nawracająco-zwalniającą na początku choroby, całkowitym lub znacznym wycofaniem się ubytków neurologicznych po pierwszym rzucie, długim okresem remisji między dwoma pierwszymi rzutami oraz długim okresem między pierwszym rzutem, a uzyskaniem 4 punktów w skali Kurtzkego. Zła prognoza jest związana z wieloogniskową symptomatologią pierwszego rzutu i wystąpieniem objawów uszkodzenia układu piramidowego, znaczną częstością rzutów w czasie pierwszych 5 lat choroby i szybkim narastaniem niesprawności w pierwszych 5 latach choroby.
Niestety do tej pory nie wynaleziono leku, który wyleczyłby ze stwardnienia rozsianego. Leczenie sprowadza się więc do próby wpłynięcia na przebieg choroby oraz łagodzenia jej skutków. Wiele leków jest nadal w fazie badań klinicznych, a ich skuteczność nie jest w pełni udowodniona. Problem stanowi również ograniczona dostępność do znacznej części leków powodowana ich wysokim kosztem.
4 grupy leków stosowane w farmakoterapii SM to:
1) Leki immunomodulujące. Mają za zadanie zmienić naturalny przebieg choroby, opóźnić jej rozwój poprzez zmniejszenie liczby rzutów choroby i ich nasilenia. Wczesne podanie leku daje szansę na zahamowanie procesu chorobowego i zmniejsza ryzyko niepełnosprawności. Obecnie większość leków z tej grupy jest w fazie badań dotyczących ich skuteczności oraz długofalowego działania.
2) Leki immunosupresyjne. Ich skuteczność nie jest w pełni udowodniona (mało prób klinicznych, znaczna cytotoksyczność, w większości stanowią tzw. terapię ratunkową przy znacznym pogorszeniu stanu zdrowia pacjenta), mają działanie tłumiące układ odpornościowy.
3) Leki objawowe. W zależności od objawów, które pojawiają się u konkretnych chorych. Leki przepisuje zwykle lekarz prowadzący lub lekarze specjaliści, aby złagodzić pojawiające się objawy. Leczenie objawów oprócz farmakoterapii obejmuje również rehabilitację, fizjoterapię oraz psychoterapię.
4) Leki stosowane podczas rzutu choroby (najczęściej kortykosteroidy). Ich zadaniem jest również immunosupresja tj. tłumienie działania układu odpornościowego w celu powstrzymania procesów zapalnych. To leczenie dotyczy głównie chorych z postacią rzutowo-remisyjną SM. Warunkiem wdrożenia leczenia jest rozpoznanie rzutu (czyli pojawienia się nowych lub nasilenia istniejących objawów neurologicznych, które pogarszają stan chorego).
 Według wskazań medycznych leki immunomodulujące powinny być podawane osobie chorej już przy pierwszych objawach choroby, są one tzw. lekami pierwszego rzutu dla pacjentów z SM. Dzisiaj już wiadomo, że stosowanie interferonów beta (beta-1a i beta-1b) wpływa korzystnie immunomodulująco na przebieg schorzenia, czego wyrazem jest zmniejszenie częstości nawrotów i hamowanie postępu inwalidztwa. W niektórych krajach interferony beta podawane są natychmiast po stwierdzeniu przez chorego pierwszych objawów choroby potwierdzonych następnie za pomocą MRI. W przeciwieństwie do interferonów beta, interferon gamma, który także budził pewne nadzieje, jest cytokiną, która pogarsza przebieg SM. Został on wycofany z leczenia tej choroby. Jednak te światowe standardy są dostępne dla chorych w Polsce w stopniu marginalnym. Ważnym trendem w badaniach nad nowymi, eksperymentalnymi metodami leczenia jest terapia preparatami antygenowospecyficznymi. Należy tu wspomnieć o prowadzonych ostatnio badaniach nad bezpieczeństwem użycia szczepionki BHT-3009, zawierającej pełnej długości białko podstawowe mieliny (MBP) (wyprodukowane metodą inżynierii genetycznej), oraz jej modulującym wpływem na odpowiedź immunologiczną. Trwają badania nad stworzeniem szczepionki z użyciem receptora dla komórek T. Prowadzone są próby z terapią kombinowaną, czyli połączeniem terapii immunomodulującej i immunosupresyjnej. Aktualnie rehabilitacja w SR to interdyscyplinarna opieka nad pacjentem, obejmująca oprócz rehabilitacji ruchowej psychoterapię, przeciwdziałanie w zaburzeniach napięcia mięśni oraz problemy urologiczne, i czynności przewodu pokarmowego. Ma ona za zadanie osiągnięcie i utrzymanie maksymalnego, możliwego w danym momencie dla danego pacjenta poziomu we wszystkich sferach życia: zdrowia fizycznego i psychicznego oraz w sprawach zawodowych i społecznych. Kompleksowe postępowanie rehabilitacyjne prowadzone przez doświadczonego w neurorehabilitacji fizjoterapeutę, połączone z psychoterapią i terapią objawową jest aktualnie uznawane za najlepszy i najbardziej efektywny sposób postępowania objawowego u chorych na SR. Leczenie rehabilitacyjne powinno być modyfikowane w zależności od indywidualnych potrzeb pacjenta, służyć dostosowaniu go do jego środowiska życia, uwzględniać typ i stopień niesprawności oraz konsekwentnie realizować ustalone cele leczenia. Praktyczne zalecenia odnośnie rehabilitacji w SR są następujące: fizjoterapia musi uwzględniać indywidualne potrzeby pacjenta i być dobierana do jego objawów klinicznych, tak aby jak najdłużej zachować samodzielność szczególnie w czynnościach dnia codziennego. Zaleca się stosowanie różnych technik – usprawnianie funkcji ruchowych, poprawa koordynacji ruchów, stabilizacja i rozluźnienia mięśni, zachowanie prawidłowej postawy ciała, poprawa wzorca chodu, usprawnienie funkcji wegetatywnych. Przeciwwskazane są duże wysiłki fizyczne i ćwiczenia w okresie zaostrzenia.
Według wskazań medycznych leki immunomodulujące powinny być podawane osobie chorej już przy pierwszych objawach choroby, są one tzw. lekami pierwszego rzutu dla pacjentów z SM. Dzisiaj już wiadomo, że stosowanie interferonów beta (beta-1a i beta-1b) wpływa korzystnie immunomodulująco na przebieg schorzenia, czego wyrazem jest zmniejszenie częstości nawrotów i hamowanie postępu inwalidztwa. W niektórych krajach interferony beta podawane są natychmiast po stwierdzeniu przez chorego pierwszych objawów choroby potwierdzonych następnie za pomocą MRI. W przeciwieństwie do interferonów beta, interferon gamma, który także budził pewne nadzieje, jest cytokiną, która pogarsza przebieg SM. Został on wycofany z leczenia tej choroby. Jednak te światowe standardy są dostępne dla chorych w Polsce w stopniu marginalnym. Ważnym trendem w badaniach nad nowymi, eksperymentalnymi metodami leczenia jest terapia preparatami antygenowospecyficznymi. Należy tu wspomnieć o prowadzonych ostatnio badaniach nad bezpieczeństwem użycia szczepionki BHT-3009, zawierającej pełnej długości białko podstawowe mieliny (MBP) (wyprodukowane metodą inżynierii genetycznej), oraz jej modulującym wpływem na odpowiedź immunologiczną. Trwają badania nad stworzeniem szczepionki z użyciem receptora dla komórek T. Prowadzone są próby z terapią kombinowaną, czyli połączeniem terapii immunomodulującej i immunosupresyjnej. Aktualnie rehabilitacja w SR to interdyscyplinarna opieka nad pacjentem, obejmująca oprócz rehabilitacji ruchowej psychoterapię, przeciwdziałanie w zaburzeniach napięcia mięśni oraz problemy urologiczne, i czynności przewodu pokarmowego. Ma ona za zadanie osiągnięcie i utrzymanie maksymalnego, możliwego w danym momencie dla danego pacjenta poziomu we wszystkich sferach życia: zdrowia fizycznego i psychicznego oraz w sprawach zawodowych i społecznych. Kompleksowe postępowanie rehabilitacyjne prowadzone przez doświadczonego w neurorehabilitacji fizjoterapeutę, połączone z psychoterapią i terapią objawową jest aktualnie uznawane za najlepszy i najbardziej efektywny sposób postępowania objawowego u chorych na SR. Leczenie rehabilitacyjne powinno być modyfikowane w zależności od indywidualnych potrzeb pacjenta, służyć dostosowaniu go do jego środowiska życia, uwzględniać typ i stopień niesprawności oraz konsekwentnie realizować ustalone cele leczenia. Praktyczne zalecenia odnośnie rehabilitacji w SR są następujące: fizjoterapia musi uwzględniać indywidualne potrzeby pacjenta i być dobierana do jego objawów klinicznych, tak aby jak najdłużej zachować samodzielność szczególnie w czynnościach dnia codziennego. Zaleca się stosowanie różnych technik – usprawnianie funkcji ruchowych, poprawa koordynacji ruchów, stabilizacja i rozluźnienia mięśni, zachowanie prawidłowej postawy ciała, poprawa wzorca chodu, usprawnienie funkcji wegetatywnych. Przeciwwskazane są duże wysiłki fizyczne i ćwiczenia w okresie zaostrzenia.
Mimo iż wciąż nieuleczalne, SM nie zagraża bezpośrednio życiu. Większość chorych żyje równie długo, jak osoby zdrowe. Uznawane jest za stan chroniczny. Oznacza to, że gdy się zachoruje, ma się SM do końca życia, jednak nie zaraża się nim nikogo, ani nie przekazuje go potomstwu. Zarówno w przypadku mężczyzn jak i kobiet, SM nie wpływa na możliwość założenia rodziny i posiadania dzieci. Jeszcze do niedawna lekarze przestrzegali kobiety z SM przed rodzeniem dzieci. Obecnie, jak wynika to z badań, wiadomo, że ciąża nie zmieniają przebiegu SM, nie wiąże się bezpośrednio z ryzykiem utraty sprawności fizycznej. Nie istnieje też większe zagrożenie urodzenia dziecka niepełnosprawnego. W okresie ciąży wiele kobiet z SM czuje się bardzo dobrze i ma rzadziej rzuty. Jednakże istnieje zwiększone ryzyko rzutu w miesiącach zaraz po rozwiązaniu, ze względu na ogromny wysiłek, związany z wychowaniem niemowlęcia, jego pielęgnacją, karmieniem. Opieka nad dzieckiem, zwłaszcza tuż po porodzie, jest bowiem bardzo wyczerpująca, ponad siły kobiety z SM. Dobrze zatem, aby podejmując decyzję o posiadaniu dzieci przyszli rodzice wzięli pod uwagę konieczność wspólnej opieki a nawet zwrócenia się o pomoc do dalszej rodziny lub wynajęcia opiekunki.
Polskie Towarzystwo Stwardnienia Rozsianego jest największą i jedyną ogólnopolską organizacją oddaną wspomaganiu ludzi z SM, ich rodzin i opiekunów. Stanowi główne źródło rzetelnych informacji o SM. Reprezentuje środowisko polskich chorych na forum międzynarodowych: jako członek Multiple Sclerosis Inernational Federation oraz European MS Platform. Posiada oddziały lokalne w większości dużych miast. Telefoniczna Infolinia o SM oferuje informacje, poradnictwo i pomoc dla wszystkich osób z SM, ich rodzin, przyjaciół i opiekunów. Wszystkie telefony są traktowane z pełną poufnością. Przy telefonie dyżurują osoby z SM, które rozmawiają z setkami osób nowozdiagnozowanych. Numer 0-801 313 333 [płatność jak za połączenia lokalne].
Co druga osoba poruszająca się na wózku inwalidzkim jest chora na SM. W Polsce ok. 75% chorych przechodzi na rentę niecałe 2 lata po zdiagnozowaniu SM. W chwili obecnej zaledwie 2% spośród wszystkich chorych na SM w Polsce jest objęta leczeniem immunomodulującym refundowanym przez NFZ. W porównaniu z Polską w krajach europejskich dostępność leczenia jest na dużo wyższym poziomie. W większości krajów Unii Europejskiej leczenie ma zapewnione ok. 30% chorych, a w USA około 45%. Polska plasuje się wśród krajów europejskich, na przedostatnim miejscu pod względem dostępności do terapii. Samo leczenie też nie należy do najtańszych, miesięczny wydatek na lek immunomodulujący wynosi ok. 5 tys. zł, a dochodzą do tego jeszcze inne leki i fizjoterapia. Na terapię refundowaną w Polsce mają szanse tylko osoby młode, w wieku 18-30 lat i z tego powodu osobom w wieku ponad 30 lat odmawia się leczenia.

Dodała: Marta


























